
Elegant catalysts offer quicker routes to complex compounds
When looking for ways to build up an organic molecule, traditionally chemists ignored carbon–hydrogen bonds. Unless activated by a nearby functional group such as a carbonyl, alcohol or amine, they were deemed unreactive. Recent years have seen a shift in this thinking, with new tools being developed that attack ‘inert’ C–H bonds and replace them with bonds such as carbon–carbon, carbon–oxygen, carbon–nitrogen and carbon–halogen.
This approach to putting molecules together allows chemists to shorten and simplify routes to previously-built organic molecules, and to reach molecules that they couldn’t previously synthesise.
Discovery chemists in the pharmaceutical industry are particularly interested in these techniques as a way to make many derivatives of molecules of interest in one step.
Site and stereocontrol
Breaking a strong C–H bond is challenging, but even more so is controlling which one reacts in a typical organic molecule containing dozens of different C–H bonds. Borrowing the directing group concept from traditional organic chemistry is one approach. Another popular, and more streamlined, way is to control a catalyst’s properties to tune which C–H bond in a molecule it will react with. This is possible because these bonds all have subtly different electronic and steric properties that the right catalyst can pick out.
Huw Davies at Emory University, Atlanta, US, is one of a growing band of researchers choosing this second approach. His team has been developing a toolkit of dirhodium catalysts for selectively converting different types of C–H bonds into C–C bonds. First, he reported a system that can pick out a hydrogen on the most sterically-accessible secondary C–H bond in an alkane. Next, his team did the same trick for both tertiary C–H bonds and then primary C–H bonds.

Now, they have designed a catalyst that can selectively replace specific secondary C–H bonds on the rings in alkyl substituted cyclohexanes with C–C bonds.1 This is a particularly tough challenge because alkyl substituents offer no electronic bias to the ring, meaning there are only extremely subtle differences between the six hydrogens on the three ring-carbons furthest from the steric influence of the chain. Previous attempts achieved more modest selectivity. ‘Two years ago if you said we’d be able to distinguish cleanly between C3, C4, C5 of [an alkyl substituted] cyclohexane I would have said, “that’ll never happen”,’ explains Davies.
The catalyst, as expected, selects equatorial hydrogens over axial because they are less sterically hindered. What was surprising was the level of selectivity seen between the C3 equatorial, C4 equatorial, and the C5 equatorial. ‘In most cases we get no reaction occurring at C4,’ says Davies. They also see a strong preference for the C3 position over the C5 with yields up to 89%. The key to this selectivity is a large, elaborate, chiral but highly-symmetric catalyst into which the equatorial hydrogen at the C3 perfectly fits.
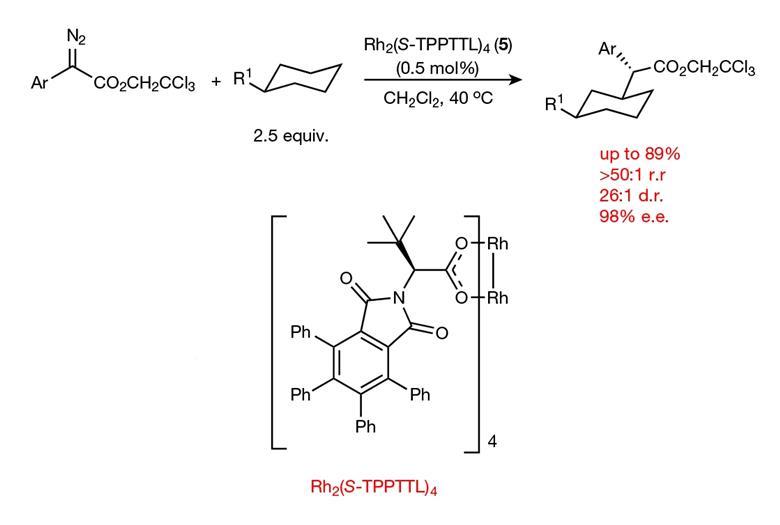
As well as excellent site selectivity, the team also report good stereocontrol. ‘We make three stereocenters,’ explains Davies. ‘Most of the reactions go with exceptionally high enantioselectivity (up to 98% ee) as well as diastereoselectivity (up to 26:1).’
Chemoselectivity
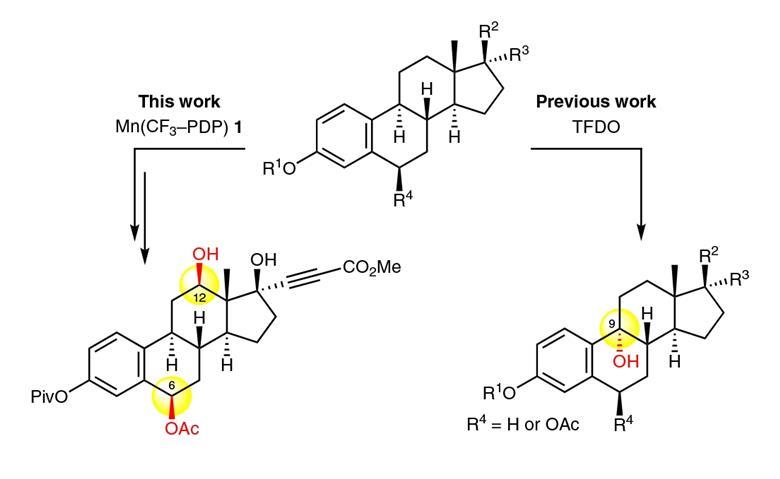
Meanwhile, Christina White at University of Illinois, US, has been designing catalytic systems that can selectively oxidise specific ‘inert’ secondary C–H bonds in molecules containing very reactive p-bonds.2 It had previously proved challenging to design oxidation catalysts with this level of selectivity.
White’s starting point was an iron catalyst system previously reported by her team that selectively attacks secondary C–H bonds in molecules containing multiple types of C–H bonds based on subtle differences in their electronic and steric properties.3 In molecules with multiple secondary C–H bonds, this catalyst favours the one most remote to any electron withdrawing groups in a molecule and any bulky centres. It struggled with chemoselectivity, however, in the presence of reactive aromatic groups.

White found that replacing the iron with a metal with a lower oxidation potential (manganese) pushed the catalyst’s preference away from the aromatic ring. Switching the ligands to bulkier ones, further favoured attacking the more easily-accessible C–H bond. A final part of the puzzle was to tune the additive that White’s signature catalytic systems use to improve yields. ‘We found we could significantly boost the reactivity, without harming chemoselectivity, by the addition of the electron-deficient carboxylic acid additive,’ she explains.
The team demonstrated the selective oxidation of a single remote secondary C–H bond, in good yields (up to 86%), in 50 different compounds containing a host of different, reactive aromatic groups regularly seen in pharmaceutical molecules. Next, the team successfully demonstrated their late-stage oxidation approach on four drug scaffolds. ‘We picked these challenging drug molecules to demonstrate the chemoselectivity in realistic settings where late stage oxidation would be desirable to create new drugs or to identify metabolites,’ White explains.
‘White and Davies are pushing further the boundaries of selectivity in the functionalisation of aliphatic C–H bonds,’ explains Olivier Baudoin, who leads a group working on C–H activation approaches at the University of Basel in Switzerland. ‘The White group’s catalyst shows unprecedented chemoselectivity for methylene oxidation on a broad range of molecules containing aromatic and heteroaromatic moieties and is therefore a major step forward to make C–H bond functionalisation methods available to a broader community of chemists. The Davies group’s catalyst displays unprecedented site- and stereoselectivity. Its ability to functionalise one particular C–H bond on these cyclic hydrocarbons is nanosurgery at its finest.’
Both White and Davies plan to make their catalyst commercially available, and say they hope others will make use of their work. ‘Hopefully this will inspire other people to think of other types of reactions that might be able to display similar types of site selectivity,’ says Davies.
References
1. J Fu et al, Nature, 2018, 564, 395 (DOI: 10.1038/s41586-018-0799-2)
2. J Zhao et al, Nat. Chem, 2018, DOI: 10.1038/s41557-018-0175-8
3 M S Chen and M C White, Science, 2007, 318, 783 (DOI: 10.1126/science.1148597)
Source
https://www.chemistryworld.com/news/new-tools-direct-reactions-at-specific-ch-bonds-in-organic-molecules/3009949.article





